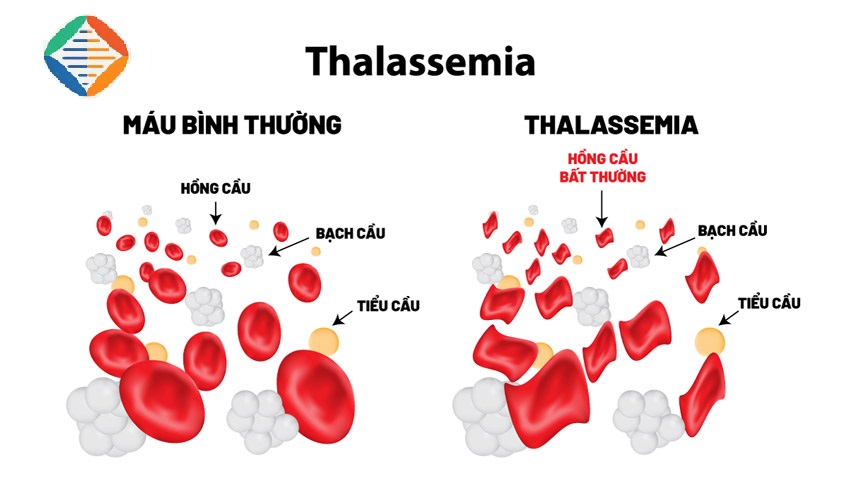
1. Tan máu bẩm sinh (Thalassemia) là gì?
Bệnh tan máu bẩm sinh (thalassemia) là một nhóm bệnh huyết sắc tố gây thiếu máu, tan máu di truyền. Mỗi thể bệnh là do bất thường tổng hợp một loại chuỗi globin; Có hai thể bệnh chính là alpha thalassemia và beta thalassemia; ngoài ra có các thể phối hợp khác như thalassemia và bệnh huyết sắc tố.
2. Nguyên nhân bệnh tan máu bẩm sinh (Thalassemia) ?
Hemoglobin gồm có 2 thành phần là Hem và globin, trong globin gồm có các chuỗi polypeptid. Bệnh Thalassemia xảy ra khi có đột biến tại một hay nhiều gen liên quan đến sự tổng hợp các chuỗi globin, dẫn đến tình trạng thiếu hụt các chuỗi globin này, làm cho hồng cầu vỡ sớm (tan máu), và biểu hiện triệu chứng thiếu máu. Bệnh nhân mắc Thalassemia có thể nhận gen bệnh từ bố hoặc mẹ, hoặc cả bố và mẹ.
Bệnh được gọi tên theo chuỗi globin bị khiếm khuyết, gồm có 2 loại bệnh Thalassemia chính:
- α-Thalassemia: Thiếu hụt tổng hợp chuỗi α, do đột biến tại một hay nhiều gen tổng hợp chuỗi α-globin.
- β-Thalassemia: Thiếu hụt tổng hợp chuỗi β, do đột biến tại một hay nhiều gen tổng hợp chuỗi β-globin.
3. Triệu chứng bệnh tan máu bẩm sinh (Thalassemia) ?
Do mất cân bằng trong quá trình tổng hợp chuỗi globin nên dẫn đến việc sinh hồng cầu không đáp ứng được nhu cầu của cơ thể và gây ra các hậu quả:
- Thiếu máu:
Là tình trạng thiếu máu mạn tính trong suốt cuộc đời người bệnh. Nguyên nhân do các chuỗi globin thừa lắng đọng trong các tế bào đầu dòng hồng làm quá trình sinh hồng cầu không hiệu lực từ trong tủy xương, hồng cầu trưởng thành bị tiêu hủy sớm hơn ở lách và lượng huyết sắc tố trong mỗi hồng cầu thấp. Tất cả các nguyên nhân này dẫn đến lượng huyết sắc tố của bệnh nhân thalassemia thấp hơn bình thường.
- Thay đổi cấu trúc xương:
Do thiếu máu, cơ thể phản ứng bằng tăng sinh hồng cầu, mở rộng diện tích sinh hồng cầu trong tuỷ xương dẫn đến thay đổi cấu trúc xương sọ, mặt và đầu xốp các xương dài, một số trường hợp có các u sinh máu (sinh máu trong ống tuỷ, phổi…). Điều này làm gương mặt bệnh nhân thalassemia bị biến dạng: trán dô, mũi tẹt, gò má cao, răng vẩu, xương dễ gãy, giảm mật độ xương, loãng xương.
- Lách to:
Chuỗi globin thừa tạo thành thể vùi trong hồng cầu làm hồng cầu mất độ mềm mại, dễ bị bắt giữ tại lách, làm lách phì đại, với một số lượng lớn hồng cầu được giữ trong lách làm giảm lượng hồng cầu trong máu do đó càng làm máu bị loãng hơn. Nếu lách bị cắt thì hiện tượng này sẽ xảy ra đối với gan.
- Rối loạn chuyển hóa sắt:
Tuỷ xương tăng sinh hồng cầu kích thích cơ thể tăng hấp thu sắt từ đường tiêu hóa, bên cạnh đó do bệnh nhân thường xuyên phải truyền khối hồng cầu nên tổng lượng sắt của cơ thể tăng cao nhanh chóng. Khi sắt huyết thanh tăng 10 – 15 lần, các vị trí gắn sắt của transferrin đã bão hoà hết, sắt sẽ gắn không đặc hiệu với các chất khác như albumin, citrate, aminoacid và được lắng đọng tại các tổ chức như gan tim, tuyến nội tiết làm tổn thương các cơ quan này. Điều này dẫn đến bệnh nhân bị xơ gan, suy gan, suy tim, suy các tuyến yên tuyến sinh dục, đái tháo đường, suy giáp, suy cận giáp…
- Rối loạn đông cầm máu:
Người bệnh tan máu bẩm sinh có những biến đổi về đông cầm máu, nhìn chung có xu hướng tăng đông.
4. Đường lây truyền bệnh tan máu bẩm sinh (Thalassemia) ?
Bệnh Thalassemia không phải là bệnh truyền nhiễm như bệnh viêm gan virus, lao phổi,.. Mà bệnh di truyền do bệnh nhân nhận gen bệnh từ cả bố và mẹ. Do đó, bệnh Thalassemia là một bệnh có tính chất gia đình.
5. Các biện pháp điều trị bệnh tan máu bẩm sinh (Thalassemia)?
Điều trị thiếu máu
- Chỉ định: Bệnh nhân có chỉ định truyền máu khi 2 lần kiểm tra đều cho thấy Hb<7g/dl, hay Hb>7g/dl nhưng có biến dạng xương.
- Truyền máu: Bệnh nhân được truyền hồng cầu lắng, máu mới, lượng 10ml/kg trong 2-3h. Tần suất truyền máu có thể mỗi 4-6 tuần một lần tùy mức độ. Bệnh nhân cần được theo dõi chặt chẽ trong quá trình truyền máu tại bệnh viện.
- Mục đích: duy trì Hb>10g/dl, giúp trẻ phát triển bình thường, tránh biến dạng xương.
Điều trị ứ sắt
- Chỉ định thải sắt được đặt ra ở trẻ trên 3 tuổi, khi mà ferritin huyết thanh >1000ng/ml
- Mục đích: ngăn ngừa tổn thương các cơ quan do ứ sắt, đặc biệt là tim và nội tiết.
- Desferoxamin tiêm dưới da 30-50mg/kg trong 8-12h × 5-7 ngày/tuần, hoặc tiêm tĩnh mạch lúc truyền máu.
- Chú ý theo dõi thị lực, thính lực hàng năm.
Cắt lách
- Chỉ định: Thalassemia thể nặng, cường lách (lách to, giảm 3 dòng tế bào máu, lượng hồng cầu lắng truyền >250ml/kg/năm).
- Biến chứng sau cắt lách: Nhiễm trùng, tắc mạch.
Điều trị hỗ trợ
- Vitamin C
- Vitamin E
- Acid folic
Ghép tủy xương
Là phương pháp hiện đại cho kết quả tốt trong điều trị Thalassemia. Tuy nhiên, ghép tủy xương có hạn chế là khó tìm được người cho tế bào gốc phù hợp.
Theo dõi
- Hemoglobin 1 tháng/lần.
- Chiều cao, cân nặng 3 tháng/lần.
- Ferritin 6 tháng/lần.
- Kiểm tra tim mạch, nội tiết, tai, mắt; tính lượng máu truyền, vấn đề thải sắt 1 năm/lần
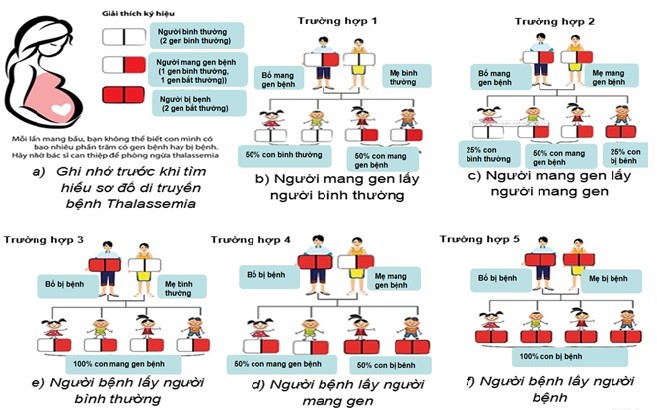
6. Vậy các bạn phải làm gì để tránh bệnh Thalassemia cho con bạn?
- Tầm soát phát hiện bệnh sớm: Tư vấn tiền hôn nhân, đặc biệt là những người thuộc nhóm đối tượng nguy cơ của bệnh cần được tư vấn, làm các xét nghiệm để được phát hiện bệnh Thalassemia sớm. Nếu cả 2 vợ chồng đều mang một thể bệnh Thalassemia, nên được tư vấn trước khi có ý định mang thai.
- Sàng lọc trước sinh, đặc biệt là đối với các cặp vợ chồng cùng mang một thể bệnh Thalassemia có thai, nên làm các xét nghiệm tầm soát và chẩn đoán gen đột biến khi thai được 12 – 18 tuần. Phương pháp được thực hiện có thể là chọc ối hoặc sinh thiết gai nhau để tìm đột biến gen (nếu có).

Hy vọng với những kiến thức trên sẽ giúp các chị em phụ nữ có thể chủ động hơn trong việc khám, sàng lọc hội chứng Thalassemia cho thai nhi. Bên cạnh đó là nên khám thai theo đúng lịch khám, không nên bỏ qua thời điểm tốt nhất để phát hiện nguy cơ dị tật bẩm sinh của trẻ trong thời kỳ mang thai. Để từ đó có những can thiệp kịp thời tránh những nguy cơ đáng tiếc có thể xảy ra.
Cần được sự tư vấn tận tình hãy liên hệ ngay với chúng tôi
Dich vụ tư vấn Vivagen: 0935.519.688

Bình luận về bài viết này